#9 – février 2023
-
Le parcours de soins
-
- En particulier, 51 postes de chargés de parcours génomique ont été déployés pour toutes les préindications du PFMG2025. Ils bénéficient d’un financement de la DGOS pour une durée de 3 ans qui élargit ainsi la mesure initiale menée dans le cadre du PNMR3. Répartis sur l’ensemble du territoire, les chargés de parcours génomique sont en charge de former et d’accompagner les prescripteurs, de contribuer à mettre en place au niveau local, puis à fluidifier, les parcours de soins des différentes préindications.
- Dans le champ des maladies rares, des RCP-FMG-MR Génomiques ont été mises en place sur l’ensemble du territoire à l’issue d’une phase pilote de 6 mois. Ces RCP-FMG plurithématiques permettent de valider la prescription d’un séquençage pangénomique pour l’ensemble des préindications maladies rares quand la situation médicale du patient respecte des critères phénotypiques de prescription, conduisant ainsi à une simplification du parcours de soins.
- En cancérologie, deux phases pilotes ont été mises en œuvre pour envisager l’utilisation de prélèvements fixés (FFPE) et d’ADN tumoral circulant. Les résultats sont attendus en 2023 et conduiront à définir les modalités de recours aux différents types de prélèvements selon les préindications.
 Chaque mois, les LBM-FMG SeqOIA et AURAGEN transmettent un bilan d’activité à l’équipe de coordination du PFMG qui en fait une synthèse à l’échelle nationale.
7 387 prescriptions ont été validées en RCP-FMG d’amont en 2022 :
Chaque mois, les LBM-FMG SeqOIA et AURAGEN transmettent un bilan d’activité à l’équipe de coordination du PFMG qui en fait une synthèse à l’échelle nationale.
7 387 prescriptions ont été validées en RCP-FMG d’amont en 2022 :
- 6 255 pour les maladies rares
- 69 pour l’oncogénétique
- 1 063 pour les cancers
- 4 248 pour les maladies rares
- 1 002 pour les cancers
-
Les projets pilotes du PFMG2025
-
- Les inclusions dans le projet DEFIDIAG, portant sur la déficience intellectuelle, se sont achevées en avril 2022. 1275 trios ont été inclus et les résultats de séquençage sont en cours d’analyse.
- 344 patients ont été randomisés dans l’essai MULTISARC, portant sur les sarcomes, avec un accès possible ensuite à 10 thérapies ciblées dans le cadre de l’essai clinique. Les inclusions dans le bras olaparib/durvalumab ont été suspendues en novembre 2022, le nombre requis de patients ayant été atteint.
- Les inclusions dans le projet en population générale POPGEN sont terminées, avec plus de 10000 participants de la cohorte Constance. Les génotypages sont achevés et le séquençage du génome de 4000 de ces participants est en cours.
- Le protocole de GLUCOGEN, portant sur le diabète, est finalisé et sera soumis très prochainement aux autorités réglementaires.
-
La réutilisation des données pour la recherche
-
- 8 projets de recherche ont déposé une demande d’accès aux données auprès du Comité Scientique et Ethique (CSE) du CAD, 5 dans le champ des maladies rares et 3 en cancérologie,
- 5 demandes d’accès aux données sont en cours d’évaluation,
- et 3 projets de recherche ont été validés par le CSE.
Le PFMG2025 a comme ambition d’intégrer la médecine génomique dans le parcours de soins des patients. 70 préindications ont été validées à ce jour (60 préindications pour les maladies rares, 2 pour l’oncogénétique et 8 en cancérologie) et ont conduit à la prescription d’un examen pangénomique pour 15 000 patients.
Les données génomiques produites par les laboratoires LBM-FMG SeqOIA et AURAGEN ainsi que celles provenant des quatre projets pilotes du PFMG2025 (DEFIDIAG pour la déficience intellectuelle, MULTIPLI pour les sarcomes des tissus mous, POPGEN en population générale et GLUCOGEN pour les formes atypiques du diabète) ont vocation à alimenter le Collecteur Analyseur de Données (CAD), constituant ainsi une ressource nationale de données génomiques et de données cliniques associées.
Plus de 7800 comptes-rendus ont été remis aux prescripteurs, dont quasiment 6000 pour des patients atteints de maladies rares et 1900 pour des patients atteints de cancers. Ainsi, les données génomiques et cliniques de 7800 patients, et le cas échéant de membres de leur famille, sont d’ores et déjà accessibles pour la recherche. L’accès aux données de DEFIDIAG et de MULTISARC est également possible sous certaines conditions tant que ces deux essais cliniques sont encore en cours.
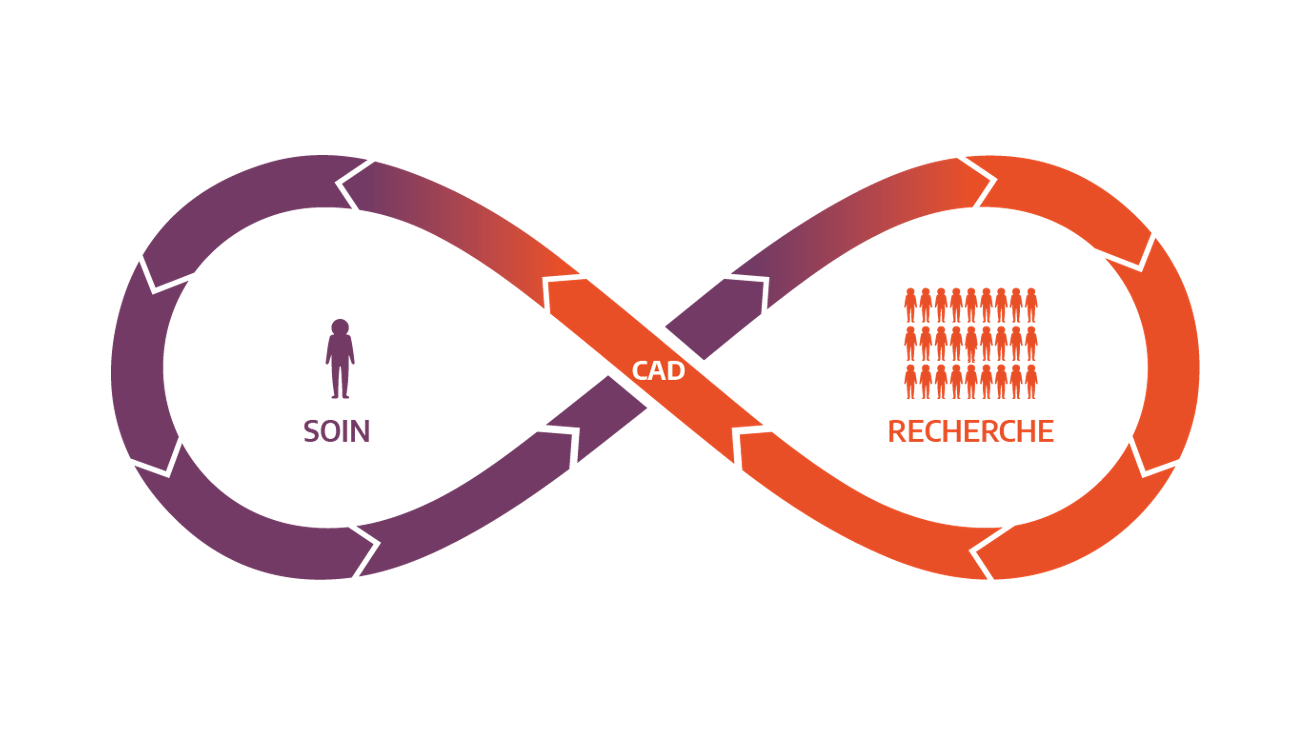 En y associant une puissance de calcul adaptée et un éventail d’outils informatiques au sein d’une infrastructure dédiée, le CAD permet d’inscrire le PFMG2025 dans un continuum entre le soin et la recherche, au bénéfice des patients. Il proposera ainsi des services pour le soin et la recherche :
En y associant une puissance de calcul adaptée et un éventail d’outils informatiques au sein d’une infrastructure dédiée, le CAD permet d’inscrire le PFMG2025 dans un continuum entre le soin et la recherche, au bénéfice des patients. Il proposera ainsi des services pour le soin et la recherche :
En y associant une puissance de calcul adaptée et un éventail d’outils informatiques au sein d’une infrastructure dédiée, le CAD permet d’inscrire le PFMG2025 dans un continuum entre le soin et la recherche, au bénéfice des patients. Il proposera ainsi des services pour le soin et la recherche :
- Dans le cadre du soin, il fournira aux praticiens des outils contribuant à l’interprétation clinico-biologique des données individuelles, ainsi qu’à leur réanalyse dans la poursuite de la démarche diagnostique ;
- Dans le cadre de la recherche, il mettra à disposition des chercheurs les données collectées dans le cadre du soin pour produire de nouvelles connaissances.
Un nourrisson est reçu en consultation de Génétique à Limoges dans les suites du diagnostic d’une surdité neurosensorielle congénitale dépistée à la naissance.
Deuxième enfant de parents non apparentés sans antécédent familial particulier, il présente une surdité bilatérale sévère à profonde, isolée, sans malformation au scanner des rochers et à l’IRM cérébrale. Cet enfant a rapidement bénéficié de la pose d’implants cochléaires. Toutefois l’évolution montre un retard de développement et d’acquisition de la marche, avec un questionnement sur d’éventuels traits autistiques. Cet enfant présente quelques particularités morphologiques avec un grand front bombé, une dystopie des canthi, une racine du nez large et une ensellure nasale marquée, à pondérer par l’origine ethnique. Ses mensurations (Taille, Poids et PC) sont dans les normes.
Les premiers bilans génétiques (analyse de DFNB1, ACPA, panel de gènes impliqués dans les surdités) reviennent sans anomalie, de même que la sérologie CMV. L’indication de séquençage du génome est alors retenue par la RCP nationale de la filière SENSGENE. Une analyse en trio (cas index et 2 parents) est réalisée dans le laboratoire AURAGEN.
Deux variations génétiques sont identifiées en trans dans le gène CDH23 :
 Le gène CDH23 est impliqué dans le syndrome de Usher de type 1 et dans la surdité non syndromique, de transmission autosomique récessive. Le syndrome de Usher de type 1 est défini par l’association d’une surdité congénitale sévère à profonde, d’une rétinopathie pigmentaire au cours de la première décade et dans la majorité des cas d’une aréfléxie vestibulaire se traduisant notamment par un retard d’acquisition de la marche.
Un phénotypage inverse pour corréler ces données génomiques aux données cliniques est en cours avec en particulier un examen ophtalmologique pour déterminer si cet enfant présente une rétinopathie pigmentaire associée à sa surdité. Il est compliqué à ce stade de donner un pronostic car même en absence de rétinopathie pigmentaire, étant donné le jeune âge du patient, on ne peut exclure l’apparition de signes ophtalmologiques plus tardifs.
Le diagnostic de syndrome de Usher à partir d’une surdité a priori isolée est utile pour adapter la prise en charge de la personne et donner un conseil génétique adéquat.
Le séquençage du génome a permis d’apporter une explication à la surdité de cet enfant alors que les analyses classiques antérieures ne l’avaient pas permis. Les outils bioinformatiques développés par le laboratoire AURAGEN ont en effet permis d’identifier un remaniement chromosomique complexe intragénique, non détecté par l’ACPA.
Même s’ils restent une cause minoritaire dans les pathologies monogéniques, les remaniements chromosomiques doivent être recherchés. Le laboratoire AURAGEN a d’autres exemples similaires de remaniements de petite taille ou d’allure équilibrée, impliqués dans les pathologies monogéniques.
Le gène CDH23 est impliqué dans le syndrome de Usher de type 1 et dans la surdité non syndromique, de transmission autosomique récessive. Le syndrome de Usher de type 1 est défini par l’association d’une surdité congénitale sévère à profonde, d’une rétinopathie pigmentaire au cours de la première décade et dans la majorité des cas d’une aréfléxie vestibulaire se traduisant notamment par un retard d’acquisition de la marche.
Un phénotypage inverse pour corréler ces données génomiques aux données cliniques est en cours avec en particulier un examen ophtalmologique pour déterminer si cet enfant présente une rétinopathie pigmentaire associée à sa surdité. Il est compliqué à ce stade de donner un pronostic car même en absence de rétinopathie pigmentaire, étant donné le jeune âge du patient, on ne peut exclure l’apparition de signes ophtalmologiques plus tardifs.
Le diagnostic de syndrome de Usher à partir d’une surdité a priori isolée est utile pour adapter la prise en charge de la personne et donner un conseil génétique adéquat.
Le séquençage du génome a permis d’apporter une explication à la surdité de cet enfant alors que les analyses classiques antérieures ne l’avaient pas permis. Les outils bioinformatiques développés par le laboratoire AURAGEN ont en effet permis d’identifier un remaniement chromosomique complexe intragénique, non détecté par l’ACPA.
Même s’ils restent une cause minoritaire dans les pathologies monogéniques, les remaniements chromosomiques doivent être recherchés. Le laboratoire AURAGEN a d’autres exemples similaires de remaniements de petite taille ou d’allure équilibrée, impliqués dans les pathologies monogéniques.
- une variation faux-sens, p.(Arg1746Gln), hérité du père, considéré comme pathogène, déjà été rapporté dans la littérature chez des patients porteurs d’un syndrome de Usher,
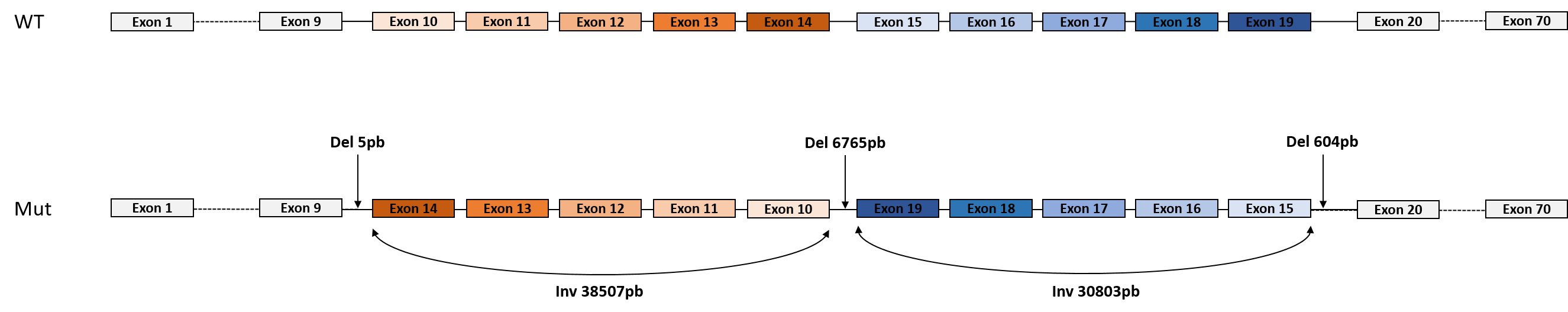
- un remaniement chromosomique complexe, hérité de la mère. Il s’agit de deux inversions côte à côte, intragéniques et associées à trois délétions introniques de part et d’autre et entre les deux inversions (cf figure). La conséquence de ce remaniement complexe est vraisemblablement pathogène et équivalente à un allèle nul. Les outils bio-informatiques du laboratoire AURAGEN ont détecté les deux inversions, mais seule la visualisation et l’analyse sur IGV par les soft-clip a permis de voir et de préciser les délétions.
Le gène CDH23 est impliqué dans le syndrome de Usher de type 1 et dans la surdité non syndromique, de transmission autosomique récessive. Le syndrome de Usher de type 1 est défini par l’association d’une surdité congénitale sévère à profonde, d’une rétinopathie pigmentaire au cours de la première décade et dans la majorité des cas d’une aréfléxie vestibulaire se traduisant notamment par un retard d’acquisition de la marche.
Un phénotypage inverse pour corréler ces données génomiques aux données cliniques est en cours avec en particulier un examen ophtalmologique pour déterminer si cet enfant présente une rétinopathie pigmentaire associée à sa surdité. Il est compliqué à ce stade de donner un pronostic car même en absence de rétinopathie pigmentaire, étant donné le jeune âge du patient, on ne peut exclure l’apparition de signes ophtalmologiques plus tardifs.
Le diagnostic de syndrome de Usher à partir d’une surdité a priori isolée est utile pour adapter la prise en charge de la personne et donner un conseil génétique adéquat.
Le séquençage du génome a permis d’apporter une explication à la surdité de cet enfant alors que les analyses classiques antérieures ne l’avaient pas permis. Les outils bioinformatiques développés par le laboratoire AURAGEN ont en effet permis d’identifier un remaniement chromosomique complexe intragénique, non détecté par l’ACPA.
Même s’ils restent une cause minoritaire dans les pathologies monogéniques, les remaniements chromosomiques doivent être recherchés. Le laboratoire AURAGEN a d’autres exemples similaires de remaniements de petite taille ou d’allure équilibrée, impliqués dans les pathologies monogéniques.
< Retour aux newsletters
Thèmes
- #Acces-aux-donnees
- #Activité
- #CAD
- #Cancers
- #Cas-clinique
- #Chargés-de-parcours-génomique
- #cpg
- #Defidiag
- #évènement
- #Interprétations
- #LBM-FMG
- #loi-de-bioéthique
- #maladies-rares
- #Malaries-rares
- #Multipli
- #Noticesinformation
- #PFMG2025
- #Préindications
- #Projets-pilotes
- #recherche
- #SHS
- 1+MG
- Activité
- erdera
- projet européen
- relations internationales
- transnational