Presentation
Most neurodegenerative diseases are genetic in origin. They may have relatively pure clinical presentations or at least be homogeneous within a family for familial forms, which allows them to be included in the other pre-indications proposed by the BRAIN-TEAM network. However, some clinical presentations, whether sporadic or familial, are more complex, combining varying degrees of highly heterogeneous clinical signs that are difficult to fit into any of the above-mentioned pre-indications. Similarly, the phenotypic spectrum of these complex entities continues to grow with the advent of new high-throughput sequencing strategies, highlighting frequent overlaps between some of these entities. It therefore seems appropriate to group these entities together under the same candidate pre-indication.
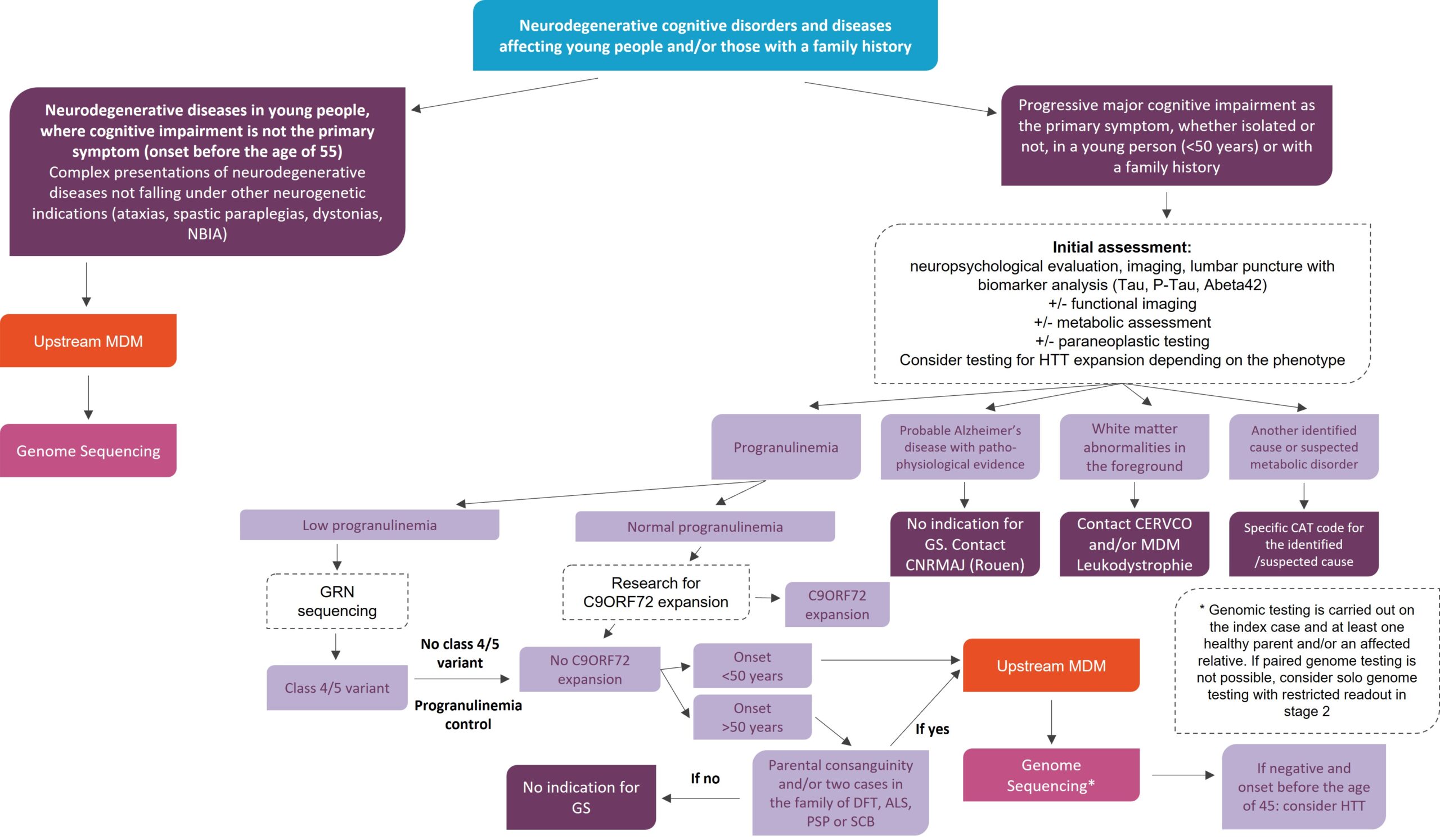
Among neurodegenerative diseases in young people, some are accompanied by progressive cognitive impairment, which may or may not be the primary symptom. The aetiological approach then follows a distinct algorithm. A significant number of genetic causes are known, some of which are less rare, such as the spectrum of frontotemporal lobar degeneration with two major causes that should be identified first (C9ORF72 expansions and GRN mutations that can be suspected after a blood test), while other diseases are ultra-rare. Early-onset Alzheimer’s disease (EAD, onset before age 65) is itself a separate entity belonging to the non-rare spectrum of Alzheimer’s disease, and has relatively low genetic heterogeneity with regard to monogenic causes, a significant proportion of EAD being non-monogenic, with a significant genetic component within a multifactorial model. Genetic analyses performed in the context of EAD are processed by the National Reference Centre for Young Alzheimer’s Disease in Rouen (www.alzheimer-génétique.fr).
Criteria before considering a discussion in MDM-FMG
Sporadic cases, onset <55 years of age, no prominent cognitive impairment
- Progressive progression
- Heterogeneous clinical symptoms encompassing different syndromic categories (pyramidal syndrome, cerebellar syndrome, Parkinson’s syndrome, peripheral neuropathy, abnormal movements (dystonia, myoclonus, chorea), leukodystrophy, neurosensory impairment, cognitive disorders, etc.) that cannot be classified under any of the pre-indications proposed by the BRAIN-TEAM network
- Etiological assessment ruling out an acquired differential diagnosis:
- Inflammatory/autoimmune; tumour/paraneoplastic
- Toxic; infectious; vascular
- Minimum assessment: MRI, lumbar puncture, neurometabolic blood and urine tests
- Possibility of sampling at least one relative
- Familial forms: analysis of at least 1 affected relative
- Sporadic forms: trio analysis or, if parents are unavailable, analysis of a parent or collateral relative over 65 years of age and free of the condition.
Major progressive cognitive impairment in the foreground, isolated or not, young subject (<50 years old) or familial
- Aetiological assessment excluding a differential diagnosis:
- neuropsychological, imaging,
- lumbar puncture with biomarkers (Tau, P-Tau, Abeta42)
- ± functional imaging ± metabolic assessment ± paraneoplastic
- Consider testing for HTT expansion depending on phenotype
- Following this assessment, in the event of:
- Probable Alzheimer’s disease with pathophysiological evidence: No indication for genome sequencing Contact the CNRMAJ (Rouen)
- Prominent white matter abnormalities: Contact the CERVCO and/or RCP leukodystrophies
- Other identified cause or suspected metabolic disease: specific course of action depending on the identified/suspected cause
In other cases, patients must have their progranulin levels measured and be tested for C9ORF72 expansion before genome sequencing is considered.
Genome sequencing is considered in cases of normal progranulin levels, absence of C9ORF72 expansion, and in patients with symptom onset before the age of 50 or, if onset after the age of 50, in cases of parental consanguinity and/or two cases in the family in the FTD/ALS/PSP/CBD spectrum.
Genome sequencing is performed on the index case and at least one healthy parent and/or affected relative. If duo genotyping is not possible, consider solo genotyping with restricted reading in stage 2.
Genome Sequencing in diagnostic strategy
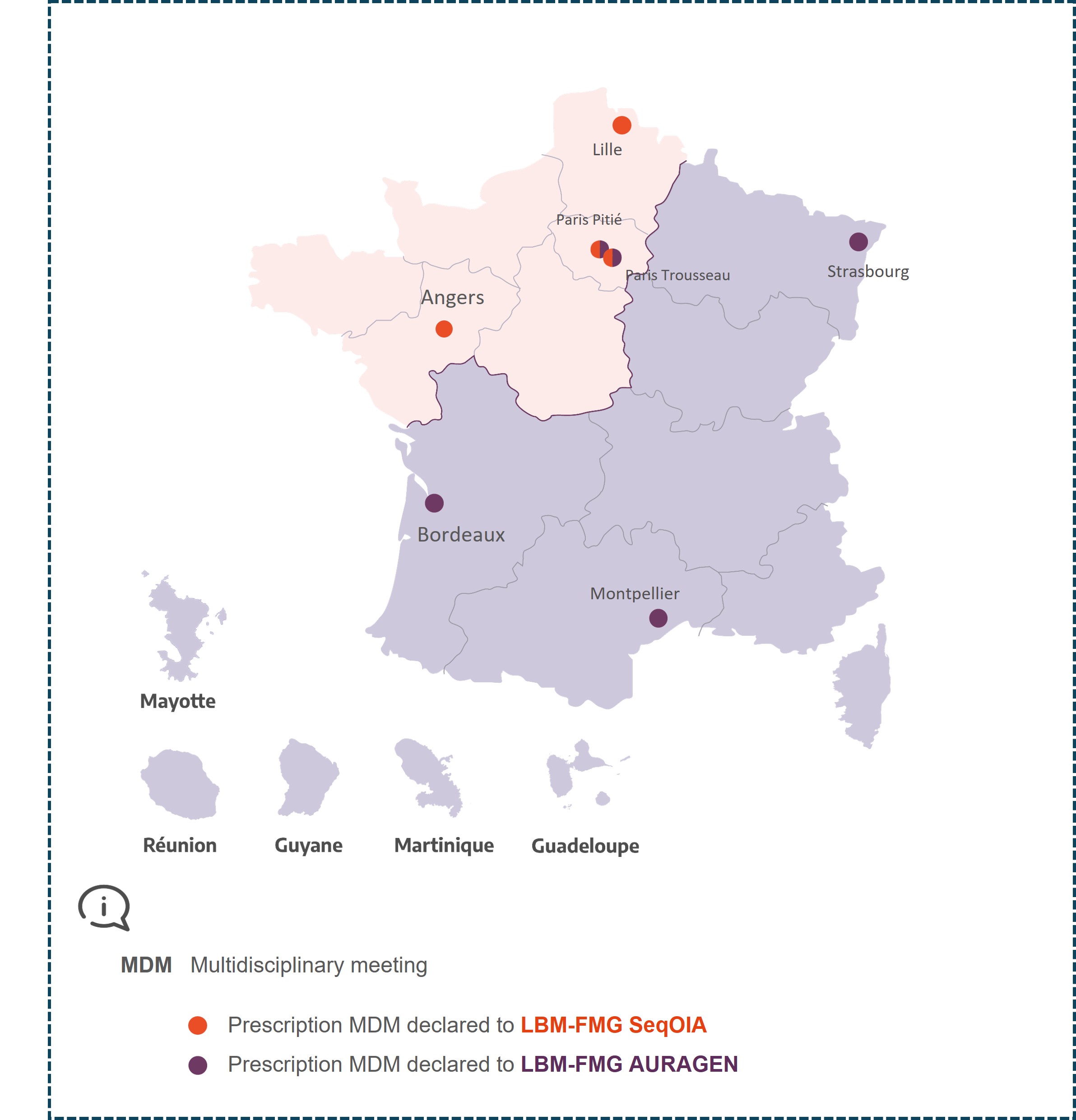
MDM cartography
MDM Neurogenetics Paris Pitié
Claire EWENCZYK
Alexandra DURR
Perrine CHARLES
Anna HEINZMANN
MDM Neurogenetics Paris Trousseau
MDM Neurogenetics Angers
MDM Neurogenetics Strasbourg
Mathieu ANHEIM
mathieu.anheim@chru-strasbourg.fr
Christine TRANCHANT
christine.tranchant@chru-strasbourg.fr
Solène FRISMAND
Matthieu BEREAU
Christel THAUVIN
Anne DOE DE MAINDREVILLE
adoedemaindreville@chu-reims.fr
Juliette Piard
MDM Neurogenetics Montpellier
MDM Neurogenetics Lille
David DEVOS
Luc DEFEBVRE
Sylvie NGUYEN-THETICH
sylvie.nguyenthetich@chru-lille.fr
Gaël NICOLAS
Eugénie MUTEZ