Presentation
Non-syndromic or syndromic pituitary deficiency (but pituitary deficiency as the primary pathology).
In the neonatal period, in the event of severe hypoglycaemia, cholestatic jaundice (corticotropic deficiency), confirmed by hormone assays; stunted growth, or syndromic form including defective pituitary development (midline defect, septo-optic dysplasia, etc.).
Later on, stunted growth or delayed puberty, particularly associated with pituitary stalk interruption syndrome (combined deficiency or isolated somatotropic deficiency).
Criteria before considering a discussion in MDM-FMG
Proven pituitary deficiency (clinical, biological, MRI) in accordance with PNDS guidelines for ‘Congenital pituitary deficiency’ September 2021.
NB: Simple stature or weight delay is not sufficient.
NB: Isolated corticotropic deficiency in adults is secondary in most cases and very rarely warrants genetic testing.
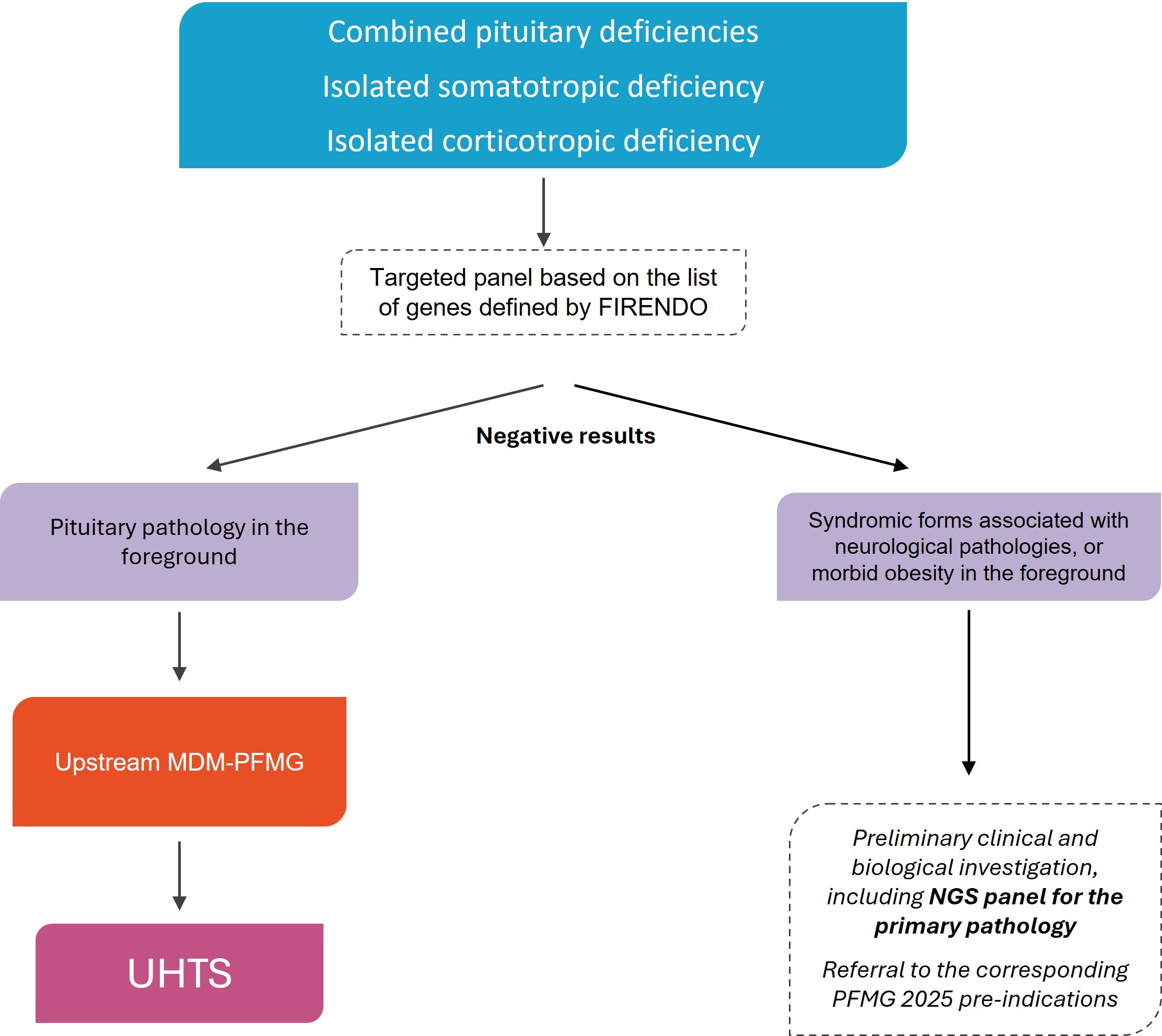
A preliminary NGS panel analysis (Firendo HAS 2023 gene list), carried out in a medical biology laboratory of reference for the pathology (JORF, Decree of 18 June 2024), is required.
NB: an exome without specification of NGS coverage/depth for genes involved in pituitary deficiencies is not sufficient).
Karyotyping and ACPA are required for syndromic cases.
NB: pituitary pathology dominating the clinical picture, see strategy.
In syndromic forms, taking into account the variable clinical expression for the same genotype, the associated phenotypes (index case and family) must be specified in detail (e.g. renal, cardiac and limb malformations (polydactyly, clinodactyly, etc.), cleft lip or cleft palate), as well as the investigations carried out.
UHTS in diagnostic strategy
MDM cartography
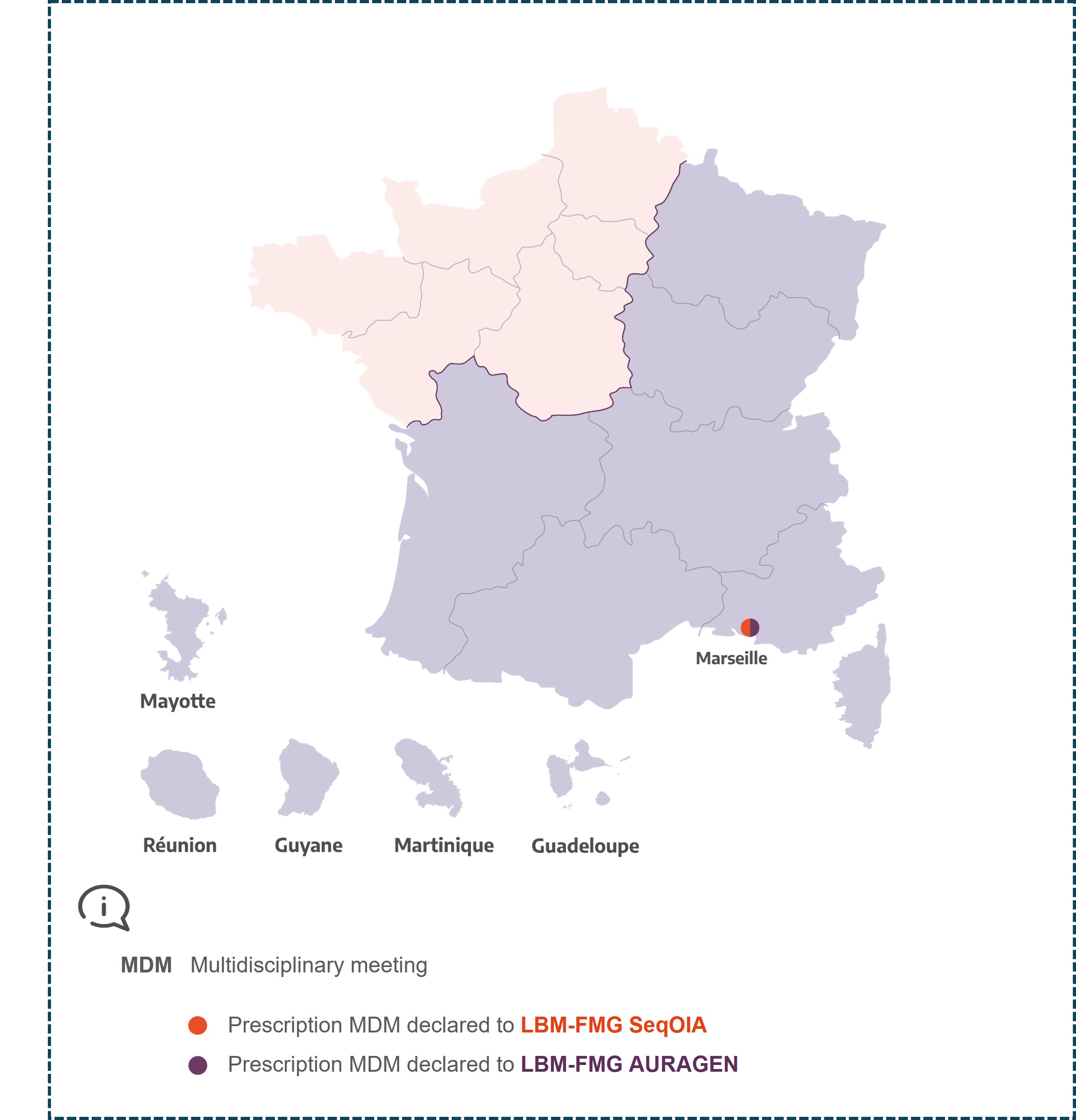
MDM Thyréo-Hypophyse
Marseille
Haifa RAHABI
For pediatric HYPO :
Sarah CASTETS